Research
Metabolic Regulation of Ferroptosis, Nutrient Dependency, and Tumor Suppression
Our lab has a long-standing interest in understanding nutrient signaling and metabolic stress response in both normal and cancer cells. We are interested in the questions: 1) how normal/cancer/stem cells sense nutrient availability? 2) how cancer cell adapt to survive and grow under metabolic stress or nutrient deprivation? and 3) how to target metabolic vulnerabilities in cancer treatment? Our previous studies focused on the role of FoxO/TSC/LKB1 tumor suppressor network in energy sensing, cancer metabolism, and stem cell maintenance. These studies have delineated an intimate link between tumor suppressor pathways that control energy sensing/metabolism and those that regulate stem cell homeostasis (Gan et al, PNAS, 2008; Gan et al, Cancer Cell, 2010; Gan et al, Nature, 2010), and identified novel mechanisms on how cancer cells adapt to metabolic stress or cancer therapy (Lin et al, 2014, Cancer Research; Liu et al, Nature Cell Biology, 2016; Xiao et al, Nature Communications, 2017; Dai et al, PNAS, 2017). Our current research focuses on two related research topics that have emerged from our more recent work: 1) the role and mechanisms of ferroptosis in cellular metabolism, tumor suppression, and cancer therapy, and 2) cystine metabolism-induced nutrient dependency and its implication in cancer therapy
1. Ferroptosis in cellular metabolism, tumor
suppression, and cancer therapy
Ferroptosis is a form of regulated cell death that is triggered
by iron-dependent lipid peroxidation with distinctive features and
underlying mechanisms from other forms of regulated cell death such
as apoptosis. Cells have evolved elegant defense mechanisms to
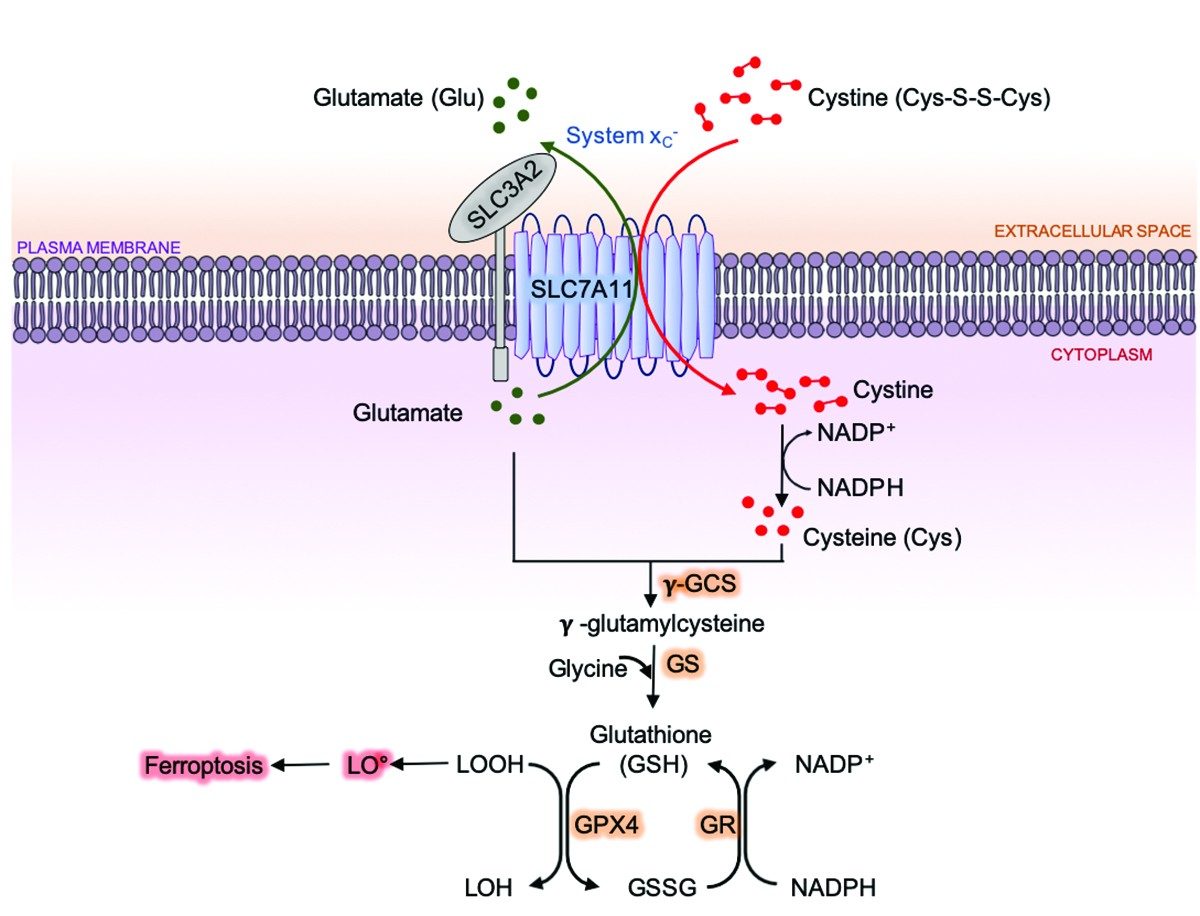
suppress ferroptosis, prominent among which is the SLC7A11-GPX4
signaling axis, wherein amino acid transporter SLC7A11 imports cystine
to generate cysteine for glutathione (GSH) synthesis and GPX4 (a
glutathione peroxidase) subsequently uses GSH to detoxify lipid
peroxides and inhibit ferroptosis (Fig. 1).
Consequently, cystine starvation or inactivation of GPX4 or SLC7A11
triggers potent ferroptosis in many cancer cells.
In recent
years, our research has delved into understanding ferroptosis in tumor
biology while we were studying tumor suppressive mechanisms of tumor
suppressor BAP1, a H2A deubiquitinase. Through comprehensive analyses
of BAP1-target genes in cancer cells, we identified cystine
transporter SLC7A11 as a key BAP1 target gene in tumor suppression and
revealed a BAP1-mediated epigenetic mechanism that links ferroptosis
to tumor suppression (Zhang et al, Nature Cell
Biology, 2018). Our subsequent study also uncovered a critical
role of ferroptosis in radiotherapy-induced cell death and tumor
suppression and suggested to combine radiotherapy and ferroptosis
inducers in cancer treatment (Lei et al, Cell
Research, 2020). Our findings together reveal that ferroptosis
is an important tumor suppression mechanism and provide a broad
framework for further understanding and targeting ferroptosis in
cancer therapy. Currently, we are employing multi-disciplinary
approaches, including sophisticated genetic mouse models, clinical
investigation, and functional studies to further dissect the role and
mechanisms of ferroptosis in tumor suppression and to therapeutically
target ferroptosis in cancer treatment.
Considering that
ferroptosis is inherently linked to metabolic stress (such as cystine
deprivation, reactive oxygen species, and iron overload), we have also
been studying the interplay between cellular metabolism and
ferroptosis. In one project, we investigated the role of energy stress
in ferroptosis regulation. While it is well known that energy stress
depletes ATP and induces cell death, we recently showed that energy
stress potently suppresses ferroptosis by activating the energy sensor
AMPK. Functional and lipidomic analyses revealed that AMPK inhibits
ferroptosis through phosphorylating acetyl-CoA carboxylase and
suppressing polyunsaturated fatty acid biosynthesis. This study
therefore reveals an unexpected coupling between ferroptosis and
AMPK-mediated energy sensing signaling (Lee et al, Nature Cell
Biology, 2020). Currently, we are applying integrated
approaches, including metabolomic, lipidomic, and proteomic analyses
and CRISPR screens, to gain deeper mechanistic understanding of
ferroptosis and its interplay with cellular metabolism.
2. Cystine metabolism-induced nutrient dependency and its
implication in cancer therapy
SLC7A11-mediated cystine uptake is critical for maintaining
redox balance and protecting cells from ferroptosis, and SLC7A11 is
frequently overexpressed in cancers. We recently made unexpected
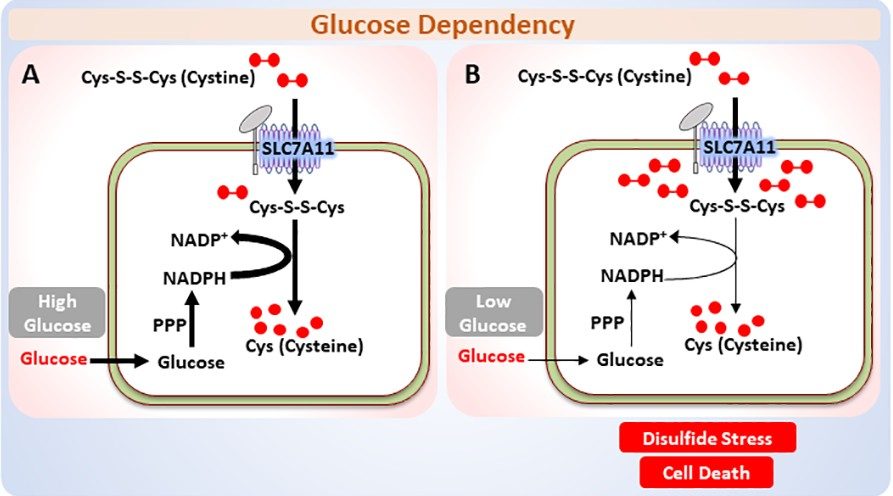
findings that this metabolic reprogramming also comes at a significant
cost for SLC7A11-high cancer cells: constitutively reducing cystine to
cysteine presents a substantial drain on the cellular NADPH pool and
renders such cells highly dependent on pentose phosphate pathway (PPP)
and glucose for survival (Fig. 2A). Limiting glucose
supply to SLC7A11-high cancer cells results in disulfide stress and
rapid cell death (Fig. 2B). Our subsequent
preclinical studies validated the concept to target this metabolic
vulnerability in SLC7A11-high cancers (Liu et al, Nature
Cell Biology, 2020). Recently we also showed that glucose
starvation-induced NADPH depletion in SLC7A11-high cancer cells can
trigger signaling events to decrease H2A ubiquitination and activate
ER stress gene expression to mediate subsequent cell death (Zhang et al, Cancer
Research, 2020).
Our future plan in this project
includes: 1) to therapeutically target SLC7A11-induced nutrient
dependency and disulfide stress in SLC7A11-high cancers, such as
BAP1-mutant renal cancer and KEAP1-mutant lung cancer; 2) to further
study SLC7A11-induced cell death under other metabolic stress
conditions; 3) to employ CRISPR screens and other approaches to
understand the nature and dissect the mechanisms of SLC7A11-induced
cell death under glucose starvation; and 4) to study disulfide
stress-initiated cellular signaling in SLC7A11-high cancer cells by
conducting redox proteomic analyses.